CCP-EM software suite v2: Doppio

Linux
CCP-EM Doppio 1.5.0 for Linux (2/2/2026)
CCP-EM Doppio 1.6 is coming soon! A beta release for Linux is available now (23/4/2026)
Mac
CCP-EM Doppio 1.5.0 for Mac (2/2/2026)
CCP-EM Doppio 1.6 is coming soon! A beta release for Mac is available now (23/4/2026)
Doppio is a web-based user interface for CCP-EM and RELION software. Please start by reading the user guide for instructions on installing and using Doppio.
Previous versions of Doppio are available here.
Changes are listed in the release notes and help with problems is available in the troubleshooting section.
Doppio is free of charge for non-profit use under the CCP-EM academic software licence. For-profit users should please contact ccpemlicensing@stfc.ac.uk for information on commercial licencing. Further details are available on the licensing page.
If you find any problems or have suggestions for useful features, please report them to the CCP-EM team at ccpem@stfc.ac.uk.
Quick links:
CCP-EM Pipeliner
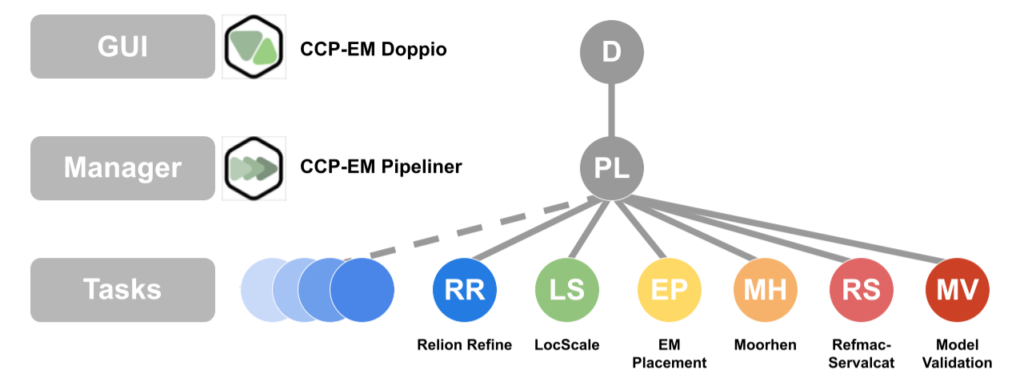
The CCP-EM Pipeliner is a Python library which provides the ‘business logic’ layer for the CCP-EM software suite (v2). It can be used be separately and is open source (MPL 2.0). Its Python API allows the design of custom, automated cryoEM workflows for SPA, STA and model building and is used as part of eBIC/DLS’s automated processing pipeliner and was also used in the CZII CryoET Object Identification Kaggle challenge. Pipeliner projects are fully compatible with Doppio and RELION. They can be viewed and continued in the Doppio UI and custom job types can also be added. For installation and more information please see GitLab repository.
CCP-EM software suite v1
The previous “version 1” CCP-EM software suite is no longer actively developed but is still available for download. A small number of jobs in Doppio need programs from the CCP-EM v1 package.
Prerequisites
| Program | Required For | Available From |
|---|---|---|
| Relion | The CCP-EM download comes with a copy of Relion 4.0 for convenience, but it is preferable to make a separate standalone installation of Relion 5.0. | https://relion.readthedocs.io/en/latest/Installation.html |
| CCP4 | CCP-EM uses programs from CCP4 for a number of tasks. To use those tasks, install CCP4 and activate it before running CCP-EM. CCP-EM is developed using the latest update of CCP4. You can manage updates via the CCP4 update manager – run ccp4um.Note that CCP4 is separately licensed. | http://www.ccp4.ac.uk/download/ |
| package-ccpem2 | Some tools are not yet included in the main CCP-EM download, but have Doppio task interfaces. package-ccpem2 will install some of these tools: checkmysequence, emda, modelcraft, tempy, locscale, emdbva, modelangelo, slicendice | https://gitlab.com/ccpem/package-ccpem2 |
| Modeller | FlexEM and some other programs require Modeller to be installed and for users to have obtained a valid licence key. Please register for a Modeller licence key (free for academic use). | Mac: Please download and install the Modeller Mac package and use default installation location. Linux: After running ./install_ccpem.sh you will be prompted to run ./install_modeller.sh. Please run this and enter the license key when prompted. |
| XQuartz | XQuartz is required for CCP-EM on Macs. Please install separately. We recommend XQuartz-2.8.2 however macOS 10.14 (Mojave) only works with XQuartz-2.8.0. This may apply to other older macOS versions. | XQuartz |
Operating Systems
CCP-EM for Linux is built on Scientific Linux 6.10. We expect this to work on all Linux versions released since approximately 2010. It is tested on Scientific Linux 6.10, Scientific Linux 7.5, Red Hat Enterprise Linux 6.10, Ubuntu 16.04 and Ubuntu 18.04.
CCP-EM for Mac is built on OS X 10.14 using the 10.9 SDK, and tested on OS X 10.11 and later.